Service de soins
Service de soins
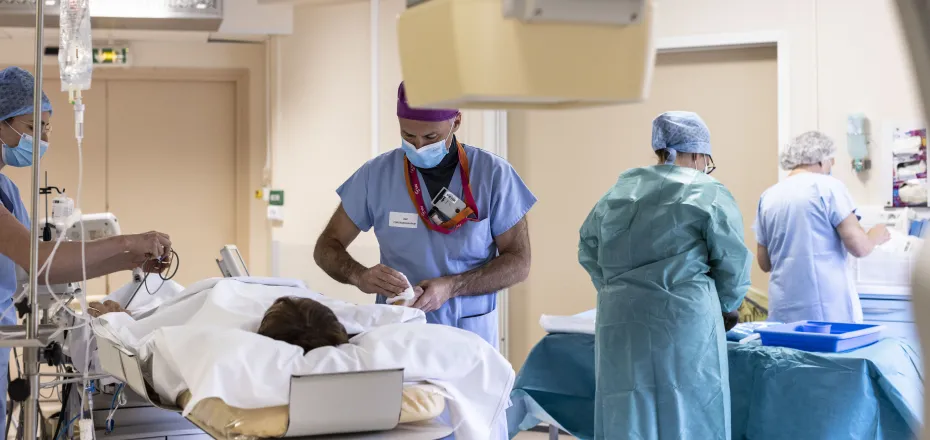
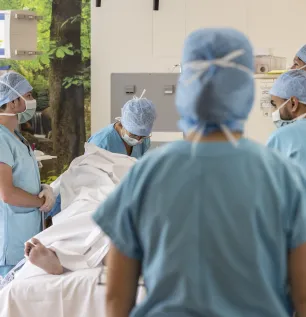
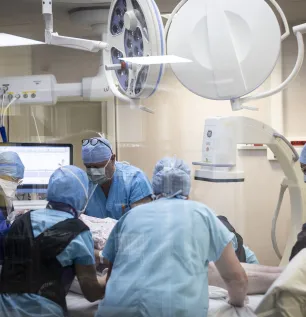
Blocs opératoires

Service de soins
Cardiologie et explorations cardio-vasculaires

Service de soins
CATTP - Hôpital de jour - Echovie

Service de soins
CeGIDD - Centre de dépistage des IST

Service de soins
Cellule d'urgence médico psychologique (CUMP)
Service de soins
Centre d'accueil et de crise (CAC)
Service de soins
Centre d'accueil thérapeutique à temps partiel - Le Bosquet

Service de soins
Centre d'expertise autisme adultes (CEAA)
Service de soins
Centre de consultations et thérapies familiales
Service de soins
Centre de ressources pour les intervenants auprès d'auteurs de violences sexuelles (CRIAVS)

Service de soins
Centre de santé sexuelle

Service de soins
Centre de soins d'accompagnement et de prévention en addictologie (CSAPA)
Service de soins
Centre de vaccination anti-amarile et de conseils aux voyageurs

Service de soins
Centre de vaccination public (CVP)
Service de soins